Strategic Divestments: Maximising Product Lifecycle Value
Published Oct 21, 2024
Published 20th December 2023

Pharmaceutical interest for synthetic peptides has been significantly revived over the last decade thanks to new production and analytical technologies. The number of clinical trial applications and marketing authorisation applications for synthetic peptides has indeed increased over the last few years. Synthetic peptides are at the interface of small molecules and biological products and therefore their quality requirements were typically not covered by existing regulatory guidelines.
This gap has now been addressed by regulatory agencies. The FDA published two new general chapters: USP <1503> (Official date: 01-Aug-2021) addressing specific quality considerations for synthetic peptide drug substances and USP <1504> (Official date: 01-Dec-2023) providing recommendations on the minimum quality attributes for starting materials used in the manufacture of synthetic peptides. In October 2023, a draft guidance on the Development and Manufacture of Synthetic Peptides has been issued by the EMA for public consultation to cover synthetic peptides with more than 4 amino acids (tetrapeptides and below are considered as small molecules). This blog explores the main quality requirements specific to this type of product, and highlights any differences observed in the two regions.
Peptides are natural or artificially manufactured short chains of two or more amino acids covalently linked by an amide bond. The exact size at which a chain or chains of amino acids become a protein is unclear. The FDA, however, distinguishes proteins from peptides based on size where any alpha amino acid polymer with a specific defined sequence that is greater than 40 amino acids is considered to be a protein.
Solid-phase peptide synthesis (SPPS) is the most common manufacturing method used, especially for larger peptides. SPPS involves the successive addition of protected amino acid (AA) derivatives to a growing peptide chain immobilised on a solid please (resin). The steps include removal of the N-terminus protecting group of the AA added, wash, coupling of the activated incoming AA, capping by acetylation and wash. Liquid-phase peptide synthesis (LPPS) is also used as a manufacturing method especially for small peptides or as part of a hybrid approach where peptide fragments are manufactured by SPPS and condensed by LPPS. USP <1503> highlights that the using SPPS or LPPS will lead to differences in the impurity profiles. Chromatographic techniques, are generally used as purification methods.
The EMA draft guideline provides information on the level of detail expected in the relevant manufacturing CTD sections. In particular, for SPPS, the final deprotection step should be described in detail in Section 3.2.S.2.2. The use of scavengers and other reagents should also be included in this section. Examples of critical steps to be included in Section 3.2.S.2.4 are also provided in the EMA draft guideline. These includes the deprotection step, control of washing steps, coupling or capping reaction monitoring, control of cleavage steps, drying steps. For the purification step, the pooling strategy of the individual fractions of the preparative chromatography should also be addressed in this section.
Special attention must be paid to the quality attributes of the starting materials (SM) as they have a direct impact on the impurity profile of the drug substance (DS). The general monograph USP <1504> focuses specifically on the quality criteria for the SM used in the manufacturing with emphasis on impurities. The requirements of ICH Q11 and associated Q&A documents are applicable to synthetic peptides. Protected amino acid derivatives (AAD) are generally acceptable as SM and commonly include the below quality attributes:
Related impurities could originate from the contaminants of unprotected AA (AA enantiomers, diastereoisomers, foreign AAs) or from the AAD manufacturing process (unprotected AA, dipeptides, ꞵ-alanyl impurities). Other impurities may include residual solvents, water content and elemental impurities.
The EMA draft guideline states that information, in the form of flowcharts, indicating the synthetic process(es) of all starting materials including details of reagents, solvents and catalysts used, should be provided in Section 3.2.S.2.3, followed by a criticality assessment of which starting material impurities may have an impact on the impurity profile of the peptide.
Additional components may be conjugated to the peptide to alter or enhance the properties, for example, conjugation to poly(ethylene glycol) (PEG), lipids and proteins has been used as a half-life extension strategy. Conjugation can also be used to deliver a cytotoxic payload or imaging agent to specific cell types targeted by the peptide. In these cases, the conjugate or its precursor should be identified as an additional starting material. The choice and control of this starting material should be justified as laid down in ICH Q11 and its associated Q&A.
As always, a list of all other materials (i.e. solvents, chromatographic materials) used in the manufacturing process is expected in Section 3.2.S.2.3. The resin used in SPPS is not considered as a SM unless it is preloaded with the first AA.
The structure of the peptide should be confirmed by analytical data. This includes the primary, secondary, tertiary and quaternary structure, where relevant. Table 1 summarises common tests and analytical techniques used for synthetic peptide structural characterisation.
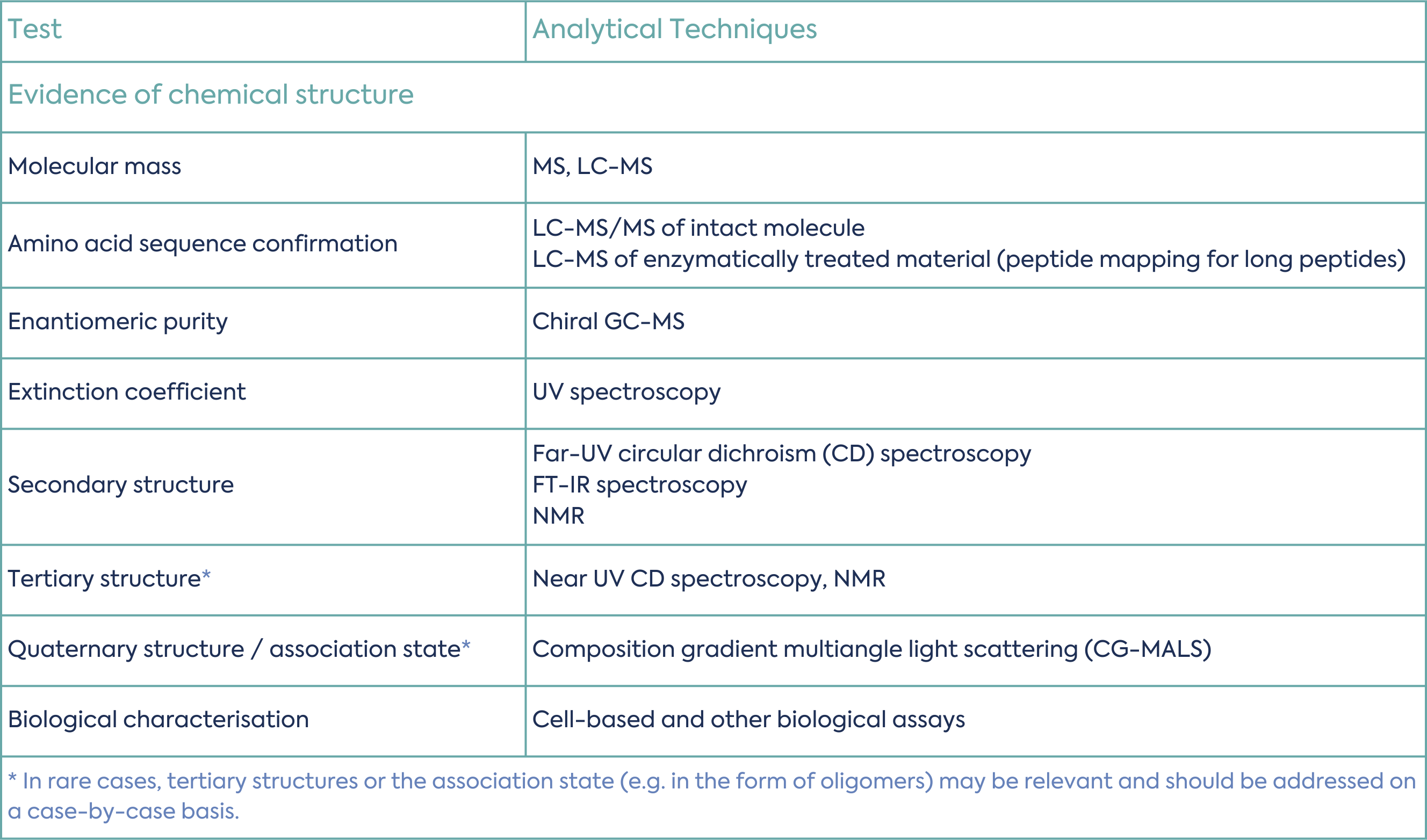
Purity is one of the most important critical quality attributes (CQA) for synthetic peptides.
Peptide related impurities can originate from the SMs, be formed during the synthesis (due to undesired or incomplete reactions during SPPS or cleavage) or arise upon degradation during storage. The impurity profile is typically highly complicated due to the structure and to the risk of co-eluting impurities, and analytical development may include several different methods used in combination and a careful evaluation of mass balance.
Peptide related impurities formed during the manufacturing process may include:
Degradation impurities can be the result of oxidation, hydrolysis, deamidation, diketopiperazine and pyroglutamic acid formation, β-elimination, condensation and formation of dehydropeptides and disulfide cleavage/exchange. The degradation pathway should be evaluated during forced degradation studies taking into account the amino acids composition and sequence: e.g. oxidation of Cys and Met residues, deamidation, hydrolysis, β-Asp-containing sequences.
The general chapter USP <1503> includes information on the common analytical methods used, such as LC-MS, LC-MS/MS or HPLC/UPLC, depending on the type of peptide related impurities.
Non-peptide impurities include process reagents, by-products, residual solvents and elemental impurities. Any residuals of reagents and/or solvents should either comply with ICH M7 (if genotoxic), or ICH Q3C/ ICH Q3D thresholds, or be toxicologically qualified.
Synthetic peptides are excluded from the scope of ICH Q3A “Impurities in New Drug Substances”. The EMA draft guideline confirms that the limits laid down in ICH Q3A are therefore not applicable and the specific thresholds for peptide-related impurities are defined in the Ph. Eur. general monograph ‘Substances for Pharmaceutical Use’ and summarised below:

It is however stated in the USP <1503> that other regulatory authorities, including the FDA, may not recognise the Ph. Eur. identification threshold of > 0.5% and may instead consider the limits for unspecified impurities on a case-by-case basis.
A parameter for each specified identified impurity, each specified unidentified impurity, any unspecified impurity, and the total impurities (as the sum of all impurities) should be included in the synthetic peptide specification.
Typical specification tests are summarised in Table 3 (non-exhaustive list):
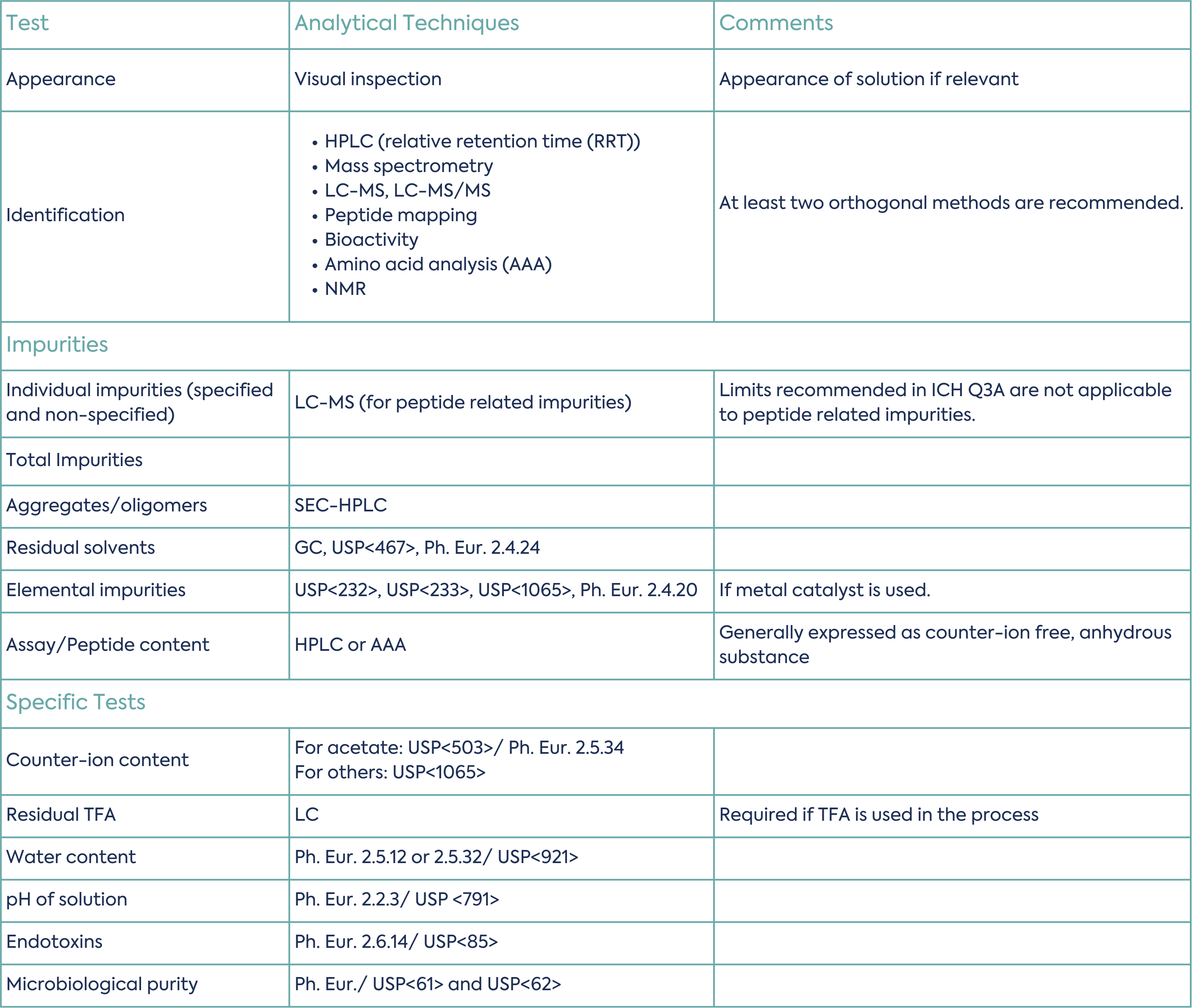
The EMA draft guideline lastly includes some information specific for medicinal products based on synthetic peptides. Here, again, the quality requirements fall between those of small molecules and biologics, and the guideline brings some clarity. The guideline reminds that requirements for risk evaluation and control of elemental impurities and nitrosamines apply. Formulation–specific considerations include whether there is interaction with the excipients, the risk of aggregation, and the importance of understanding whether the mode of action is based on primary or secondary/tertiary structure. If only primary, a potency assay of the finished product may be avoided.
Peptides are typically administered as parenteral products and the requirements of the Guideline on the Sterilisation of the Medicinal Product, Active Substance, Excipient and Primary Container should be followed for the manufacturing process, prioritising terminal sterilisation if feasible.
Chemically synthesised peptides fall outside the legal framework for a biosimilar application. Nevertheless, in cases where a synthetic peptide is developed that is intended to show therapeutic equivalence to a biological medicine, the basic principles to demonstrate biosimilarity should be considered: high similarity in terms of structure, biological activity and efficacy, safety and immunogenicity. This is outlined in the EU guideline. Similarity may be demonstrated through analytical comparability testing, comprising physicochemical (structural) and biological (functional) assays and conventional analytical testing. It is important to note the stipulation in the EU guideline that, unlike a standard biosimilar application, the reference product must be sourced from the European market.
In the US, a synthetic peptide drug product that refers to a previously approved peptide drug product of recombinant deoxyribonucleic acid (rDNA) origin can be submitted as an abbreviated new drug application (ANDA) or a new drug application (NDA), depending on its impurity profile as compared to the impurity profile of the reference peptide.
The gap in regulatory requirements for synthetic peptides, which fall outside the requirements of small molecules or biologics has recently been addressed by regulatory authorities with, for example, the issue of the new EMA draft guideline and USP monographs. These guidance documents provide clarification on the starting materials selection and control, the manufacturing process, the characterisation, the impurity profile and the specifications of synthetic peptides.
Additionally, the EMA draft guidelines include some information specific for synthetic peptides medicinal product applications using biologic products as reference products. In the US, this is addressed in a separate guideline.
If you are interested in exploring regulatory services for synthetic peptides and ensuring compliance with the latest guidelines, get in touch with our expert team. Navigating the evolving landscape of regulatory requirements demands a comprehensive understanding. Our regulatory services offer tailored support in addressing CMC considerations, impurity profiles, and specifications for synthetic peptides. Contact us to discuss how we can assist you in meeting regulatory standards and facilitating a smooth development and approval process for your synthetic peptide products.


Published Oct 21, 2024

Published Oct 18, 2024

Published Oct 10, 2024

Published Oct 07, 2024

Published Sep 26, 2024

Published Sep 24, 2024

Published Sep 10, 2024

Published Aug 30, 2024
Published Aug 19, 2024