Innovating Non-Clinical Testing: NAMs and ESG Impact
Published Jul 21, 2025
Published 24th September 2024
The purpose of this blog is to provide guidance for navigating the current and proposed EU legislation regarding the incentives available for paediatric development from the completion of a Paediatric Investigational Plan (PIP) and how this may be impacted if the indication of a medicinal product also benefits from having an orphan designation in the EU. Both data exclusivity and paediatric rewards are important tools for Marketing Authorisation Holders (MAHs) to be able to extend the protection of innovative medicines, thus increasing their commercial value. Therefore, being able to employ a strategy to take advantage of these incentives is critical. In addition, the new paediatric requirements as a result of the new legislation is also outlined.
In order to incentivise paediatric development for new active substances, the Paediatric Regulation (EC 1901/2006) provides for the possibility to extend a Supplementary Protection Certificate (SPC) of a product for 6 months on the completion of an agreed PIP if the following conditions are met:
The application for the extension of the SPC should be submitted by a company at least 2 years in advance of the SPC expiry date.
The SPC extension allows for the intellectual property rights of the MAH for the product to be extended, therefore providing an additional monopoly to compensate for the time taken to receive the MA.
To note, neither a product-specific waiver nor class waivers for the approved indication allow for this paediatric incentive. Nonetheless, the paediatric incentive can still be received if the results of a completed PIP do not support a paediatric indication.
However, if you have completed a PIP for a product with an orphan designation, a different reward is available, which is an additional 2 years of marketing exclusivity. Under the Orphan Regulation (EC) 141/2000, an approved product with orphan designation benefits from 10 years of marketing exclusivity, and therefore the additional 2 years for a completed PIP takes this to 12 years in total.
The marketing exclusivity for an orphan medicine means that an application for an MAA or extension to an MAA can’t be accepted for the same therapeutic indication in respect of a similar medicinal product, unless;
The notion of a similar product means that the product has a similar principle molecular structure, the same mode of action and covers the same indication. Marketing exclusivity runs parallel to the other data exclusivity and market protection that applies to new active substances. This protection essentially prevents other manufacturers with generic versions or alternative formulations from entering the market.

It is not currently legislatively possible for an MAH with an orphan medicinal product to choose which of the incentives under the paediatric regulation (i.e. 6 months SPC extension or 2 additional years of marketing exclusivity) to benefit from. This can therefore cause a predicament if the MAH would benefit most from the 6 months SPC extension due to the timing of the SPC, as per the example below.
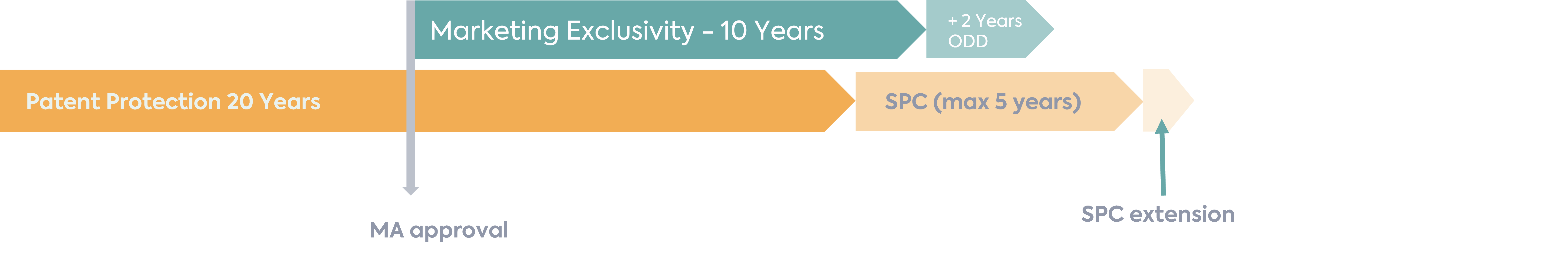
In this instance, despite not being within the spirit of the orphan legislation, it is possible to withdraw the orphan designation before the filing to include the paediatric indication or data to the MA in order to benefit instead from the extra 6-month SPC extension instead. This mechanism has previously successfully been used by the DLRC group.
However, if a company wishes to withdraw the orphan designation to take advantage of the 6 months SPC extension, careful consideration should be taken with other benefits that are granted to orphan products which may also be of use as outlined in our recent orphan article. If you would like to ensure that your orphan and paediatric strategy is carefully considered under the current legislation, then Orphix is here to assist.
In April 2023, the European Commission (EC) proposed changes to the current EU pharmaceutical legislation, marking a significant reform. This reform consists of two proposals: one directive and one regulation. The European Parliament (EP) has made amendments to some of the proposed changes in April 2024. Following this, the EC’s position will be adopted, and trilogues (negotiations between the EC, EP, and Member States) are expected in Q1 or Q2 of 2025, followed by a second reading and final adoption.
As part of this new legislation, paediatric and orphan incentives and paediatric requirements in general were revisited to ensure that the new legislation is fit for purpose given scientific advances and to continue to be attractive to pharmaceutical developers to develop medicines in the EU. More information on the changes specifically relating orphan medicines can be found in our orphan blog post.
The main incentive for completing paediatric studies in compliance with a PIP will remain the same in the form of a 6-month SPC extension. According to EC slides, this will also apply to orphan medicines (i.e., the current separate reward of two years market exclusivity for paediatric indications of orphan products will no longer apply) and therefore will benefit more companies who will no longer have to withdraw the orphan designation to take advance of this incentive.
This is expected to yield to an increased number of medicinal products, in particular, in areas of unmet medical needs of children, which are expected to reach children faster than today. It would also ensure a fair return of investment for medicinal products developers who fulfil the legal obligation to study medicinal products in children, as well as reduced administrative costs linked to the procedures that follow from the obligation.
For existing products to be exclusively developed for use in children, which are no longer covered by an SPC or a patent that qualifies as a SPC, a paediatric-use marketing authorisation ‘Paediatric Use Marketing Authorisation (PUMA)’ offers 6 years (7.5 years is proposed by the European Parliament) regulatory data protection.
Paediatric medicines will also qualify for various incentives from both the EU and Member States aimed at boosting research, development, and availability of these vital products. However, the specifics of these incentives are still to be decided.
The draft regulation is based on the existing system of obligations and incentives laid down in the Paediatrics Regulation (EC) No. 1901/2006. The draft Directive expands the Paediatric Obligation to cover:
(Note: Biohybrids are elaborate formulations or devices that integrate biological principles and methods with artificial systems for drug delivery and therapeutic applications, offering biocompatibility, environmental responsiveness, and targeted functionalities.)
There is also room for temporary waivers of the Paediatric Requirements during public health emergencies. This applies to medicinal products meant for treating, preventing, or diagnosing serious or life-threatening diseases directly related to the emergency.
The current Paediatric Committee (PDCO) will be dissolved. The expertise of the PDCO is retained and will be reorganized in the form of working parties and a pool of experts. EMA can choose to consult the Committee for CHMP or other working parties before deciding on a PIP, but this is optional. Representation of patients and health care professionals with expertise in all areas, including rare and paediatric diseases, will be increased at the Medicinal Products for Human Use (CHMP) and Pharmacovigilance Risk Assessment Committee (PRAC).
Under the new draft Regulation, the EMA might impose a PIP when a waiver is requested. This applies when a medicinal product targets a molecular aspect responsible for a different disease or condition in children within the same therapeutic area, even if the product is meant for adult conditions that don’t occur in children.
PIPs will need to be more detailed, specifying any measures to adapt the pharmaceutical form, strength, route of administration, and even the administration device for use in children.
The EMA, based on new information, may request modifications to a PIP during its assessment. Even after a PIP is agreed upon, the EMA can still ask for changes.
Except in special cases, PIPs (or waiver applications) must be submitted to the EMA before starting safety and efficacy clinical studies in adults. Late submissions could result in financial penalties.
If a PIP is discontinued, applicants need to report this to the EMA at least six months in advance and provide reasons. The EMA will make this information public. Failure to notify the EMA about discontinuation plans could also lead to financial penalties.
To ease the timing of PIP submissions, applicants will be able to submit an “initial” PIP in the following cases:
An initial PIP will detail the proposed measures to assess the quality, safety, and efficacy of the medicinal product that is currently known i.e. a high-level plan, at the time of submission. It must also include precise timelines for when updated PIPs and the final PIP will be submitted to the EMA. Missing these deadlines could result in a financial penalty.
Deferrals will have a specific time limit, initially set to a maximum of 5 years. This period can be extended in justified cases, but any extension can’t exceed another 5 years.
Product or class waivers for diseases and conditions that only occur in adults will be granted only when a Paediatric Investigation Plan (PIP) can’t be applied for based on the product’s or class’s mechanism of action for another disease or condition in children. This is to ensure more medicines are studied for paediatric use, aligning the EU regulations with similar US requirements. EMA will maintain a public list of all waivers granted.
The EMA can review and revoke an already granted waiver at any time. If a waiver is revoked, the Paediatric Requirement will be paused for 36 months from the date it is removed from the EMA’s list of waivers.
There will be a two-year deadline to market a product with a paediatric indication, starting from the date the paediatric indication is authorised;
As an EU Small, Medium Enterprise (SME), Orphix is keeping a close eye on the changes proposed by the new EU legislation and how this impacts our clients.
Based on the current orphan European pharmaceutical legislation, the incentive of completing a PIP differ when the product is an orphan medicinal product. In this case, the incentive is a two-year extension to the already granted ten years of marketing exclusivity.
This is in comparison to non-orphan medicinal products that can benefit from a six month extension to the SPC for 6 months. As these incentives are mutually exclusive, MAH’s need to come up with innovative solutions in order to maximise the benefits from the paediatric and orphan benefits by potentially withdrawing the orphan designation, despite not being in the spirit of the orphan legislation.
As proposed in the EU pharmaceutical legislation reform slides provided by the European Commission, (The EU Pharmaceutical Reform (europa.eu)), the potential 6-month SPC extension will also apply to orphan products, which then negates an MAH from needing to withdraw the orphan designation to make use of this incentive.
For general consideration of the coming new legislation, the overall regulatory strategy with regards to paediatric development should be developed prospectively considering:
If you need guidance or support on the incentives of EU paediatric development and how to navigate the new proposed pharmaceutical legislation, DLRC are here to help you at every step of the way to make sure that you’re prepared for all eventualities. Contact our team of regulatory experts via the link below, or email us at hello@dlrcgroup.com.


Published Jul 21, 2025

Published Jul 11, 2025

Published Jul 07, 2025

Published May 29, 2025

Published May 29, 2025

Published May 29, 2025

Published May 29, 2025

Published May 01, 2025

Published Apr 28, 2025